
Archived Content
The National Institute of Mental Health archives materials that are over 4 years old and no longer being updated. The content on this page is provided for historical reference purposes only and may not reflect current knowledge or information.
Turning on Dormant Gene May Hold Key for Correcting a Neurodevelopmental Defect
Finding Shows Therapeutic Potential of Small-Molecule Targeting Strategy
• Science Update
Scientists working in cell culture and in mice have been able to correct the loss of gene activity underlying a rare but severe developmental disorder by turning on a gene that is normally silenced in brain cells. Further testing of the identified compound that activates the gene will determine whether it has potential as a genetically-based treatment for the disorder, Angelman syndrome.
Background
Infants with Angelman syndrome appear normal at birth, but show developmental delays by 6 to 12 months. Features of the disorder include impaired speech, seizures, hyperactivity, and motor difficulties. No effective treatment exists.
In the late 1990s, researchers found that the disorder results from changes or deletions in the maternal gene for the enzyme ubiquitin protein ligase E3A (Ube3a). Most genes are inherited in sets of two, one from the mother and one from the father. In some cases, either the maternal or paternal gene is silenced, or prevented from being translated into protein. This normal silencing based on inheritance from a mother or father is called imprinting. The Ube3a gene is an example of genetic imprinting, as the paternal gene is normally silenced in neurons. With the maternal gene out of action, infants with Angelman syndrome lack the enzyme, leading to changes in the brain that underlie the symptoms of the disorder.
This Study
This research is reported online in the journal Nature, and was carried out by scientists at the University of North Carolina School of Medicine at Chapel Hill, led by the labs of Ben Philpot, Bryan Roth, and Mark Zylka. In an effort to restore the absent Ube3a enzyme in neurons, the research team screened thousands of compounds for their ability to “wake up” the paternal Ube3a gene. The investigators used neurons from genetically engineered mice to test whether compounds could activate the gene; the neurons fluoresce if the paternal Ube3a gene is expressed, or translated into protein. The team screened 2,306 candidate small molecules from multiple molecular libraries. If fluorescence was detected, that meant that the test compound activated the Ube3a gene. The screening and access to the molecular libraries was made possible through NIMH’s Psychoactive Drug Screening Program, funded by contract to perform pharmacological and functional screening of novel compounds. Bryan Roth at UNC Chapel Hill is the project director and a coauthor of the Nature paper.
The investigators found that a class of compounds—topoisomerase inhibitors—could unsilence the paternal gene. They chose one, topotecan, and tested it to see whether it could do the same thing in vivo in a mouse. They administered topotecan directly into the brain and later into spinal fluid; in both cases it was able to activate paternal Ube3a. Activation persisted for 12 weeks after delivery of the compound had stopped.
Significance
"This is the first time anyone has used a small molecule to successfully target activation of a disease-relevant gene," said senior author Benjamin Philpot. "The work demonstrates that turning on a dormant gene could represent a therapeutic intervention for Angelman syndrome."
NIMH helped to fund this project and has issued a grant to the UNC team to continue studies of topotecan, initially in mice. Although topotecan is already in use in both children and adults as a cancer chemotherapeutic agent, further testing is essential to determine the dosage of the agent that would be needed to be effective, the best means of administering the medication, and whether side-effects at the necessary dosage level are within a range that make it feasible to use. The authors emphasize that much work remains before this or related agents can or should be used for treatment of this condition.
Reference
Huang, H.-S., Allen, J., Mabb, A., King, I., Miriyala, J., Taylor-Blake, B., Sciaky, N., Dutton, J. Jr., Lee, H.M., Chen, X., Jin, J. Bridges, A., Zylka, M., Roth, B., Philpot, B. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. Published online ahead of print December 21, 2011, doi: 10.1038/nature10726.
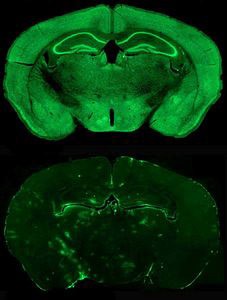
Source: Ben Philpot, Ph.D., University of North Carolina School of Medicine