
1.2 Radiotracers for Serotonergic Receptors
1 Ligand Development
1.2 Radiotracers for serotonergic receptors
Serotonin is a major neurotransmitter acting on as many as 14 different receptors. The serotonin subtype-1A (5-HT1A) receptor is one of the most extensively studied, especially because specific agonists and antagonists exist, and because it is a target for therapy in various neuropsychiatric disorders, notably depression. In 1995-1996, Dr. Pike introduced the first effective radiotracers for imaging 5-HT1A receptors in human brain, first [methyl-11C]WAY-10063513 and second [carbonyl-11C]WAY-10063514. The latter radiotracer, due to the metabolic characteristics of WAY-100635 (Ki, 0.3 nM; cLogP, 3.28; 22),15,16 is vastly superior. This has become the "gold standard" for clinical PET studies,17 even though in fact a later described radiotracer, [carbonyl-11C]-desmethyl-WAY-100635 (IC50, 1.4 nM; cLogP, 2.77; 23),18 has better imaging properties (same signal, but higher brain radioactivity per unit of administered radioactivity)19. There has been interest to have a longer-lived radiotracer; the NIH PET group were successful in developing [18F]FCWAY (Ki, 0.52 nM; cLogP, 2.76; 24) to meet this need.20 Although some bio-mathematical models have been developed for these radiotracers21,22 and they have found widespread application, there are still areas for possible improvement. Thus, many laboratories find the regular production of the carbonyl-11C labeled ligands to be daunting and would prefer access to a radiotracer through less challenging radiochemistry, such as 11C-methylation. Moreover, [carbonyl-11C]WAY-100635 is cleared rapidly from reference cerebellum, and also plasma, which exacerbate the application of either a reference tissue or kinetic model, respectively. [18F]FCWAY has the problem that it is highly defluorinated in human subjects (but not in monkey) resulting in high uptake of radioactivity as [18F]fluoride ion into bone.23 The uptake in skull is troublesome for quantifying 5-HT1A receptors in adjacent tissue (e.g. neocortex) because of "spillover" of radioactivity through the partial volume effect. We developed novel approaches to address some of these issues.
Metabolism by amide cleavage in WAY-10063524,25 demands that the radiolabel be sited on the carbonyl side of its amide bond for effective imaging.26 For this reason we attempted to block metabolism by introducing bulkier groups near the amide bond, as in a pyridinyl-N oxide (Ki, 30 pM; cLogP, 1.87; 25) and two α-C-methyl derivatives (Ki, 2.3 nM; cLogP, 3.42; 26. Ki, 0.91 nM; cLogP, 3.81; 27). Although potent 5-HT1A antagonists were found by this approach,27,28 this strategy failed to provide a superior radiotracer, as assessed by PET experiments in cynomolgus monkey.
-RWAY in rhesus monkey brain.](/sites/default/files/images/research/research-conducted-at-nimh/research-areas/clinics-and-labs/mib/sprs/images/ligand-image01.jpg)
Metabolism of [11C](-)-RWAY was initially investigated in rats. Although two less lipophilic radiometabolites appeared rapidly in plasma, LC-MS-MS study of plasma and urine samples confirmed the absence of amide hydrolysis.31 These experiments also revealed very low penetration of [11C](-)-RWAY into rat brain (76 %SUV), in stark contrast to the high penetration seen in rhesus monkey (800 %SUV).32 Pre-administration of a Pgp blocker, cyclosporin A (CsA; 50 mg/kg i.v.), to rats resulted in an almost four-fold increase in brain penetration (to ~ 300 %SUV). Under these circumstances, brain 5-HT1A receptors were clearly imaged, whereas they were unseen in the baseline experiment. CsA had no effect on plasma radiotracer concentration or free fraction, so verifying (-)-RWAY to be a substrate for rat Pgp. These results highlight the importance of efflux pumps on the brain penetration of radiotracers and also species differences in their effects. [11C](-)-RWAY gave a metabolic profile in rhesus monkey similar to that in rat. A 2-TCM (two-tissue compartment model) described the cerebral kinetics of [11C](-)-RWAY significantly better than a 1-TCM. The estimated values of the regional BP (Fig. 2) were correlated strongly between 2-TCM and MRTM (multi-linear reference tissue model) and therefore support the use of cerebellum as a reference region. We seek to discover if this radiotracer is effective in human subjects as in monkey. Consequently, we have gained FDA approval for use of [11C](-)-RWAY in human subjects under an eIND.
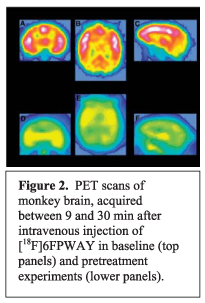
We investigated the preparation of an alternative radiotracer to [18F]FCWAY, in which the fluorine-18 label is placed in the 6-position of the pyridinyl ring of WAY-100635, namely [18F]6FPWAY (Ki, 0.33 nM; cLogP, 3.85; 29). [18F]6FPWAY was obtained by single-step nucleophilic substitution with [18F]fluoride ion33 in a synthesized bromo precursor34. After intravenous injection of [18F]6FPWAY into cynomolgus monkey, the uptake of radioactivity into whole brain reached 4.33% of injected dose at 7.5 min. Uptake was highest in 5-HT1A receptor-rich regions, reaching about twofold higher in insula than in cerebellum at 30 min. Pretreatment with WAY-100635 reduced uptake in these regions to the level in cerebellum. [18F]6FPWAY was rapidly metabolized in vivo, as evidenced by the rapid appearance of radioactive metabolites in plasma. However, importantly, there was no evidence of defluorination. [18F]6FPWAY was concluded to be moderately useful for imaging brain 5-HT1A receptors in vivo.35
![Effect of increasing dose of miconazole nitrate on rat head images obtained with [18F]FCWAY](/sites/default/files/images/research/research-conducted-at-nimh/research-areas/clinics-and-labs/mib/sprs/images/ligand-image-3.jpg)
Radiotracers having an 18Flabel attached to an aliphatic carbon, as in [18F]FCWAY, are especially prone to defluorination in vivo. We set out to investigate whether it would be possible to inhibit the cytochrome P450 enzyme (CYP2E1) that is putatively responsible for defluorination. We chose to use [18F]FCWAY with PET in a rat model. Pilot investigation in vitro suggested that miconazole might be an effective inhibitor in vivo. In baseline PET experiments, [18F]FCWAY was found to be effective for revealing 5-HT1A receptors in rat brain. Extensive defluorination was confirmed in this species by the high uptake of radioactivity seen in skull. Pre-administration of miconazole nitrate dose-dependently reduced the skull uptake of radioactivity and at the highest dose (60 mg/kg) obliterated skull uptake and greatly enhanced image quality.36 For the first time, this study demonstrates an important proof-of-principle i.e. that defluorination may be blocked in vivo with a suitable inhibitor. This principle might now be put in practice in human subjects with other more tolerable inhibitors of cytochrome P450.
The feasibility of imaging brain 5-HT4 receptors in monkey in vivo was demonstrated for the first time with [123I]SB 207710 (Kd, 37 pM; cLogP, 4.12; 30).37 The 125I-labeled version of this tracer was used to provide high quality in vitro autoradiographs of 5-HT4 receptors in post mortem human brain hemispheres.38
Except for the study of [18F]FCWAY, the above research was performed in collaboration with Prof. C. Halldin (Karolinska Institutet, Sweden) and Dr. H. V. Wikstrom (Groningen U., The Netherlands).