
Researchers Expand Understanding of Genetic Mechanisms Underlying Fragile X Syndrome
• Research Highlight
Fragile X syndrome (FXS) is a genetic disorder caused by the gene FMR1. It is the most common form of inherited intellectual disability and often co-occurs with other conditions like autism and epilepsy.
New research has unveiled major insights into genetic mechanisms underlying FXS and related disorders. The study found that the disorders involve extensive silencing of many genes that play key roles in building tissues and making the brain work properly.
The research was funded by the National Institute of Mental Health, the National Institute of Neurological Disorders and Stroke, the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and the NIH Common Fund's 4D Nucleome Program.
How do changes to the FMR1 gene lead to FXS?
The prevailing theory is that FXS involves changes to FMR1, a gene located on the X chromosome (one of two sex chromosomes in humans). The first part of the FMR1 gene is made up of repeats of a specific DNA sequence called a CGG. The CGG sequence can expand uncontrollably, leading to an excess number of repeats. A normal-length FMR1 sequence has less than 40 CGG repeats. In contrast, a mutation-length FMR1 sequence has over 200 CGG repeats.
When the FMR1 CGG reaches this mutation length, it causes physical changes to the gene that silence its expression. As a result, FMR1 produces only a little or none of its protein, which is needed for healthy brain function. This is when FXS emerges. People with FXS lacking the FMR1 protein can experience significant impacts on their development , including intellectual and learning disabilities; speech and language difficulties; and social and behavioral problems, such as hyperactivity and anxiety.
However, changes in FMR1 alone do not account for the full range of FXS symptoms, as evidenced by mouse models in which the Fmr1 gene is “knocked out,” or genetically turned off. Previous research has also implicated a broader set of genetic mechanisms and gene locations in FMR1 silencing.
What did researchers do in the current study?
Jennifer Phillips-Cremins, Ph.D. , and the study’s first authors, Thomas Malachowski, M.S., Keerthivasan Chandradoss, Ph.D., Ravi Boya, Ph.D., and Linda Zhou, M.D., Ph.D., at the University of Pennsylvania led researchers in looking beyond the standard model of FXS. They examined whether human FXS tissues show changes in the genome (the complete set of genetic material [DNA]) or the epigenome (chemical modifications that influence gene expression without altering the DNA sequence) and whether those changes are specific to FMR1 or affect other genes as well.
The researchers combined multiple high-tech analytic methods, including molecular mapping, DNA imaging, epigenetic sequencing, and genetic engineering. Additional computational approaches allowed them to integrate and find patterns in the large datasets they used.
The researchers examined genetic, epigenetic, and imaging data in multiple human cell lines from people with FXS and people without the disorder. They looked genome-wide for changes occurring on both the X chromosome and non-sex chromosomes (known as autosomes ). In addition, they worked with the NIH NeuroBrainBank to collect postmortem tissue from a brain area linked to FXS—the caudate nucleus—from people with and without the disorder.
These advanced methods enabled the researchers to look at the shape of the DNA in the entire genome and how it folds into complex 3D structures inside the cell nucleus (known as the 3D genome). The folding of the genome in 3D space reflects the packaging and interactions of segments of chromatin (the combination of DNA and protein that makes up the genome). Precise folding patterns in the 3D genome are critical for proper gene regulation and cellular function, and deviations from this structure are associated with genetic disorders like FXS as well as many cancers.

In all cell lines and tissues, the researchers compared the three versions of theFMR1 CGG repeat that can occur:
- Normal length (5–45 CGG repeats)
- Premutation length (61–199 CGG repeats)
- Mutation length (>200 CGG repeats; FXS)
By analyzing the 3D genome and chromatin changes determined by the length of the CGG repeat, the researchers could find epigenetic changes and link them to gene expression silencing and genome instability that can result in genetic disorders.
What did the results show?
The results confirmed, in cells and tissue from people with FXS, previously known changes associated with the mutation-length CGG expansion, including epigenetic changes to the FMR1 gene and silencing of FMR1 gene expression. The researchers also unexpectedly discovered more widespread changes to the DNA.
They found deposits of large pockets of a silencing form of chromatin, known as heterochromatin, that is folded too tightly and makes the gene less accessible. The heterochromatin pockets extended far beyond FMR1. On the X chromosome, the heterochromatin radiated outward to silence genes upstream of FMR1 that are essential for neural cell functions such as circuit connectivity and synaptic plasticity, which could cause learning difficulties commonly experienced by people with FXS. On autosomal chromosomes, the heterochromatin silenced multiple genes related to skin, tendon, and ligament integrity, which are clinically affected tissues in people with FXS.
The researchers also found severe misfolding of DNA and possible sites of breakage along the DNA sequence or slight expansions of other repetitive sequences within the heterochromatin. These types of higher order genome folding patterns within cells are critical for proper gene function, so the identified changes in genome structure are likely relevant to FXS beyond the local silencing of FMR1.
The Cremins Laboratory coined the term beacons of repeat expansion anchored by contacting heterochromatin, or BREACHes, to refer to the network of silenced gene areas they discovered, marked by severe chromatin misfolding and genome instability. The researchers propose that the biological significance of BREACHes is silencing genes involved in processes essential for brain function, including ones controlling synaptic plasticity that enables the brain to change in response to learning and other dynamic situations. Silencing of these genes may help explain many of the developmental differences seen in people with FXS.
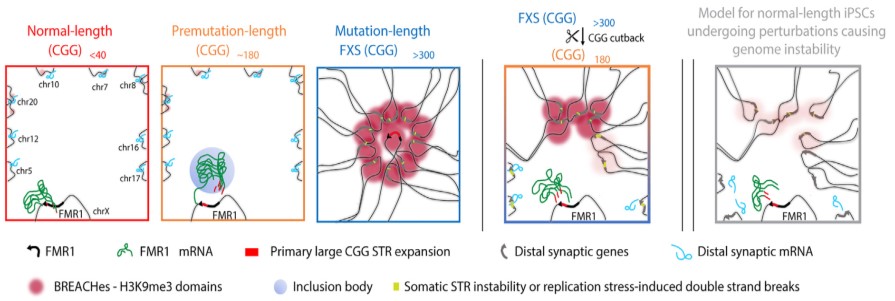
Having established broad structural and functional changes caused by the mutation-length FMR1 CGG, the researchers looked at what happened when they cut back the repeat to premutation length. They found that shortening the CGG length led to the removal of heterochromatin on many of the BREACHes. This finding indicates that the length of the CGG repeat is an important contributor to at least some of the severe and widespread 3D genome misfolding and gene silencing seen in FXS and related disorders. It also suggests that reverse-engineering the mutation-length CGG repeat could potentially prevent the emergence of genome-wide defects seen in common genetic disorders in humans.
What do the results mean?
Together, the results reveal significant changes in the genome structure related to the mutation-length FMR1 CGG sequence. These changes, associated with problems in how the DNA folds and instability in the genome, caused genes with important roles in brain function to become silent or inactive, helping explain many of the symptoms seen in people with FXS.
The silenced areas spanned a network of widespread gene locations that the researchers called BREACHes. BREACHes were observed in multiple cell types and postmortem brain tissue from people with FXS and on both the X chromosome and multiple autosomal chromosomes. BREACHes also included several genes not previously linked to FXS, offering new targets for future investigation.
According to the researchers, the discovery of BREACHes fills a major missing piece in understanding FXS. The novel finding helps explain the clinical presentation and frequent symptoms of people with FXS, which could not previously be explained by loss of the FMR1 protein alone. Because genome misfolding occurred not only in FXS cell lines but also in cells with instability in general, BREACHes might be meaningful to the growing list of disorders marked by unstable repeat expansions.
This study enhances understanding of the genetic and epigenetic mechanisms contributing to FXS disease pathology. The results may, in time, have broad relevance for understanding, diagnosing, and even treating the many disorders marked by unstable repeat expansions or genome instability, including FXS and cancer.
Reference
Malachowski, T., Chandradoss, K. R., Boya, R., Zhou, L., Cook, A. L., Su, C., Pham, K., Haws, S. A., Kim, J. H., Ryu, H.-S., Ge, C., Luppino, J. M., Nguyen, S. C., Titus, K. R., Gong, W., Wallace, O., Joyce, E. F., Wu, H., Rojas, L. A., & Phillips-Cremins, J. E. (2023). Spatially coordinated heterochromatinization of long synaptic genes in fragile X syndrome. Cell, 186(26), 5840–5858. https://doi.org/10.1016/j.cell.2023.11.019
Grants
MH120269 , MH129957 , DK127405 , DA052715 , HD098015 , NS129317